Las ataxias son enfermedades neurodegenerativas raras, muchas de ellas hereditarias, que afectan de manera especial al cerebelo y la médula espinal, y se caracterizan por una pérdida progresiva de la capacidad de coordinar los movimientos. En su mayoría son procesos irreversibles, tremendamente incapacitantes, para los que todavía no existen tratamientos eficaces.
En Europa, la ataxia hereditaria más frecuente es la ataxia de Friedreich, causada por una deficiencia de frataxina, una proteína clave para el funcionamiento de las mitocondrias, que son las encargadas de generar energía y regular el metabolismo celular. Esta deficiencia se debe a una mutación genética, en la mayoría de los casos por una expansión anómala de un triplete GAA en el ADN, que puede repetirse de 70 a más de 1.500 veces. Los síntomas suelen aparecer entre los 3 y 20 años y se manifiesta, entre otros signos, con la pérdida de coordinación motora, resultado de la degeneración de neuronas de la médula espinal y del cerebelo.
Una de las mayores dificultades para el estudio de esta enfermedad ha sido la falta de un modelo animal que imite la enfermedad, y permita estudiar el proceso neurodegenerativo a nivel molecular. Utilizando distintas aproximaciones experimentales, se han generado distintas cepas de ratones modificados genéticamente que presentaban niveles bajos de frataxina, pero que mostraban unos síntomas muy leves y resultaban, por lo tanto, poco útiles para entender la enfermedad. El hecho de no disponer de ratones que mimeticen fielmente esta enfermedad humana ha sido uno de los grandes obstáculos para la investigación sobre la ataxia de Friedreich.
Ahora, un equipo liderado por el Dr. Javier Díaz-Nido (Centro de Biología Molecular “Severo Ochoa” y Universidad Autónoma de Madrid), y por la Dra. Frida Loría (Hospital Universitario de Alcorcón), ha utilizado una nueva cepa de ratones modificados genéticamente, desarrollada por Jackson Laboratories en Estados Unidos, que sí mimetizan mejor la enfermedad humana.
Esta cepa, denominada YG8-800, fue diseñada eliminando el gen de la frataxina en ratones e introduciendo el gen humano con más de 800 repeticiones del triplete GAA. Estos ratones presentan niveles de frataxina extraordinariamente bajos y desarrollan progresivamente unos síntomas similares a la ataxia de Friedreich, con una pérdida severa de coordinación motora que se agrava con la edad. La disponibilidad de estos ratones constituye un avance extraordinario para comprender esta enfermedad y poder evaluar posibles tratamientos.
El estudio, publicado en la revista Neurobiology of Disease, aporta información valiosa sobre los mecanismos moleculares implicados en la neurodegeneración en esta enfermedad, lo que podría conducir al descubrimiento de nuevos abordajes de esta enfermedad.

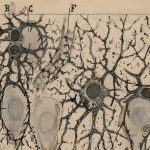
Degeneración de neuronas en el cerebelo
El análisis detallado de los ratones YG8-800 mostró a los científicos como el cerebelo se iba encogiendo (en relación con el resto del cerebro) a lo largo del tiempo. Esta atrofia progresiva del cerebelo, cuya función es crucial para la coordinación de los movimientos, puede explicar por qué los ratones van perdiendo esta capacidad con la edad. Los científicos estudiaron en detalle lo que sucedía en el cerebelo de estos ratones y se encontraron con que la atrofia del mismo se debe principalmente a la degeneración de un tipo de neuronas, las neuronas granulares en la corteza cerebelosa, que son la población neuronal mayoritaria en el cerebelo. Para entender por qué se producía esta degeneración de las neuronas en el cerebelo, los científicos realizaron toda una serie de análisis moleculares que revelaron que los ratones deficientes de frataxina presentaban unos niveles bajos de proteínas de la “cadena transportadora de electrones mitocondrial”, que funciona como la fuente de energía para las células, así como una acumulación de hierro en el cerebelo.
¿Y esto qué significa? Un mal funcionamiento de la “cadena tranportadora de electrones mitocondrial” va a provocar que disminuyan las reservas de energía que necesitan las neuronas para funcionar. Además, la acumulación de hierro puede conducir a la formación de radicales libres oxidantes, generando un proceso de estrés oxidativo que daña muchos componentes dentro de las neuronas. “Todo ello apunta al posible papel de la pérdida de función de las mitocondrias y al estrés oxidativo como factores conducentes a la degeneración de las neuronas”, afirman los autores.
Activación temprana de las células gliales
Sin embargo, los científicos se han encontrado con que lo que sucede dentro de las neuronas puede no ser lo único importante para explicar la neurodegeneración en la ataxia de Friedreich. Y es que, junto a las neuronas, nuestro sistema nervioso central contiene otras células, denominadas células de glía, que desempeñan un papel fundamental en la preservación de la viabilidad y la funcionalidad de las neuronas. Y entre estas células de glía se encuentran los astrocitos y la microglía, que también pueden ser sensibles a la deficiencia de frataxina.
“Curiosamente —mencionan los autores—, antes de detectar ningún signo evidente de degeneración neuronal, observamos la presencia de astrocitos reactivos y células de microglía activada en la corteza cerebelosa de los ratones YG8-800, que es detectable ya a los 3 meses de edad”.
¿Y en qué consiste esta “activación” temprana de las células gliales?
Para ello los científicos realizaron un análisis de las moléculas expresadas por estas células gliales, y se encontraron con un aumento en la expresión de unas proteínas denominadas citoquinas proinflamatorias, que promueven la inflamación en el cerebelo. La inflamación es una respuesta fisiológica del organismo contra las infecciones y resulta muy útil para eliminar, por ejemplo, células infectadas por virus y detener, de esta manera, la expansión del virus en el organismo. Sin embargo, cuando se dispara la inflamación de manera patológica por otra causa diferente a una infección, como en este caso de los ratones deficientes de frataxina, puede resultar en un daño a las células y así contribuir al proceso degenerativo. Además de esta respuesta inflamatoria aberrante, los autores detectaron una alteración en otras proteínas, los factores neurotróficos, que son esenciales para la supervivencia de las neuronas. “En el caso del Factor Neurotrófico Derivado del Cerebro (BDNF), observamos un incremento de su expresión a los 3 meses de edad y una marcada reducción a partir de los 6 meses”.
Estos hallazgos sugieren, en suma, que tanto la inflamación como la reducción de factores neurotróficos podrían contribuir a la degeneración de las neuronas en el cerebelo de los ratones YG8-800, que mimetizan la ataxia de Friedreich.

En definitiva, este estudio aporta luz a los posibles mecanismos responsables de la pérdida de neuronas en los ratones deficientes en frataxina. Además, abre la posibilidad de usar estos ratones YG8-800 para explorar tratamientos que puedan detener, o al menos, enlentecer, el proceso neurodegenerativo.
“Disponer de estos ratones, que imitan la neurodegeneración y los síntomas de la ataxia de Friedreich, va a ser muy útil para acelerar el desarrollo de posibles tratamientos”, concluyen los autores.
Fuente de la información: UAM Gazette
_____________________
Referencia bibliográfica:
Vicente-Acosta A, Herranz-Martín S, Pazos MR, Galán-Cruz J, Amores M, Loria F, Díaz-Nido J. Glial cell activation precedes neurodegeneration in the cerebellar cortex of the YG8-800 murine model of Friedreich ataxia. Neurobiol Dis. 2024 Oct 1;200:106631. doi: 10.1016/j.nbd.2024.106631. Epub 2024 Aug 5. PMID: 39111701.
Más acerca del Centro de Biología Molecular Severo Ochoa: https://www.cbm.uam.es/?lang=es
Más acerca da la Universidad Autónoma de Madrid:https://www.uam.es/uam/inicio
Más acerca de la revista Neurobiology of Disease: https://www.sciencedirect.com/journal/neurobiology-of-disease